Czym są chłoniaki nie-Hodgkina?
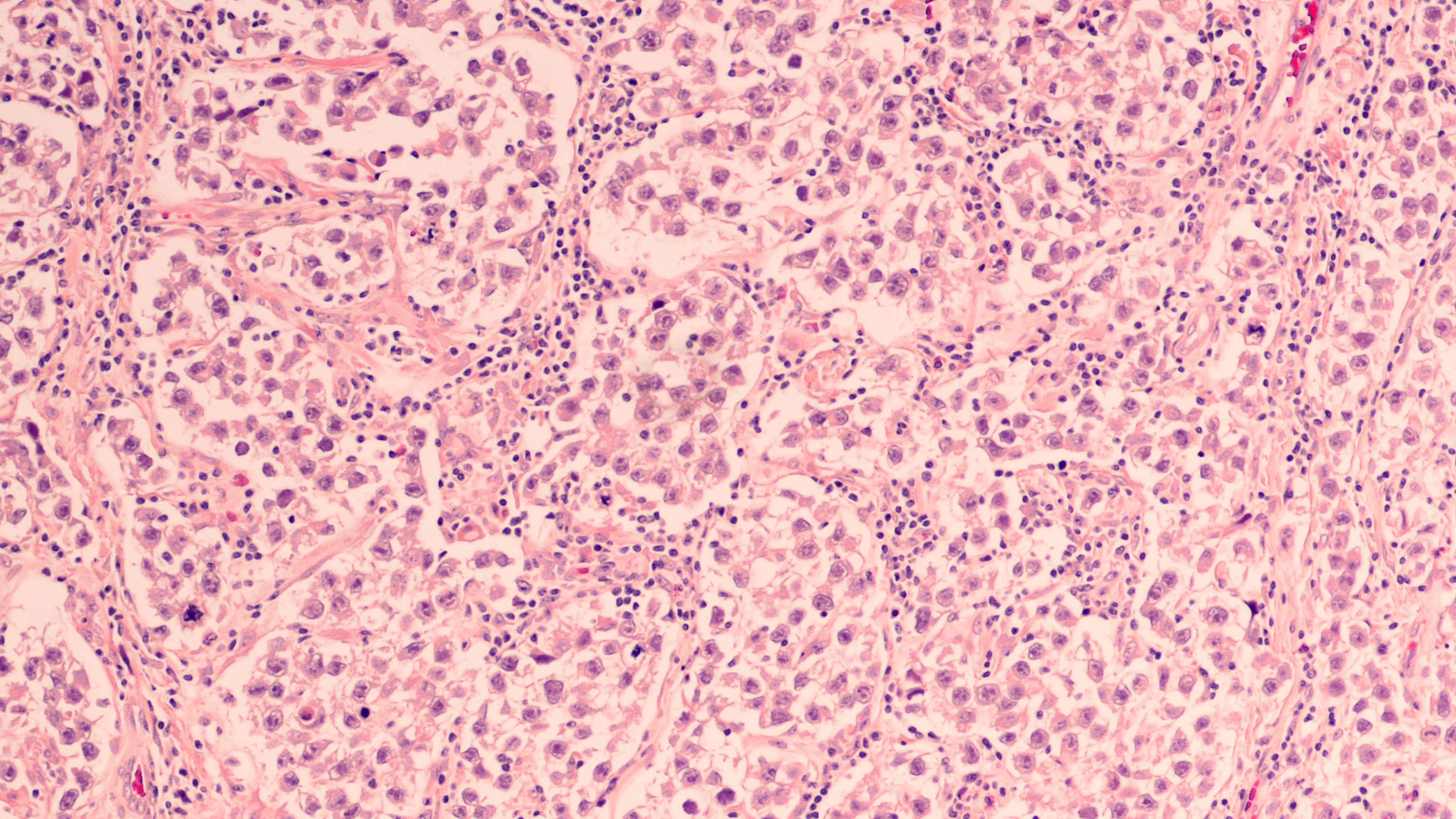
Chłoniaki są to choroby nowotworowe, w których następuje nieprawidłowy rozrost komórek układu limfoidalnego (chłonnego).
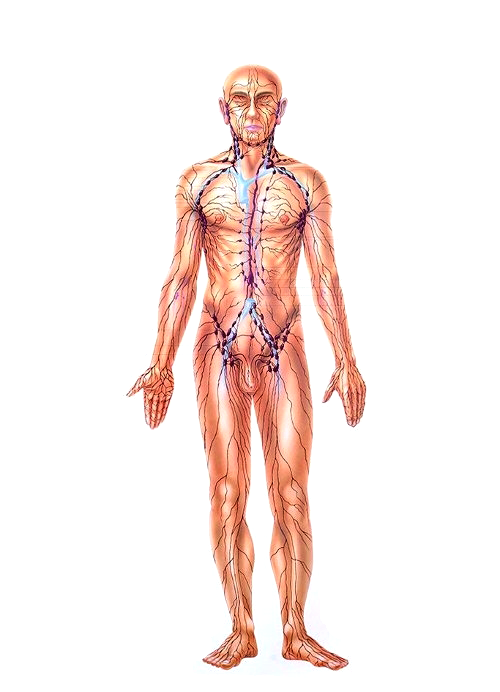
Układ chłonny człowieka jest to zespół komórek, tkanek, naczyń i narządów wyspecjalizowanych w rozpoznawaniu i eliminacji mikroorganizmów, struktur białkowych oraz komórek obcych lub własnych, ale poważnie zmienionych. Układ naczyń chłonnych ma charakter otwarty; naczynia chłonne (limfatyczne) rozpoczynają się w przestrzeniach międzykomórkowych śródtkankowych, łączą się w naczynia o coraz większym świetle, prowadzą do węzłów chłonnych, w których płyn śródtkankowy zawarty w naczyniach (chłonka) podlega filtracji (wychwytywaniu antygenów – cząsteczek mogących wywoływać reakcje odpornościowe). Naczynia odprowadzające chłonkę z węzłów chłonnych łączą się w kolejne naczynia zbiorcze, które ostatecznie tworzą przewód piersiowy mający ujście do żyły głównej górnej, gdzie chłonka wraca do układu krążenia krwi.
Do narządów limfatycznych należą, oprócz wspomnianych węzłów chłonnych, także grasica, migdałki (tkanka limfatyczna gardła tworząca pierścień Waldeyera), skupienia tkanki limfatycznej w błonie śluzowej jelit (kępki Peyera) śledziona oraz szpik kostny. Grasica jest centralnym narządem limfatycznym, w którym we wczesnym okresie życia następuje przekształcanie i dojrzewanie układu limfocytów T odpowiedzialnych za odporność komórkową (realizowaną przez komórki cytotoksyczne) . Rolę narządu centralnego dla układu limfocytów B odpowiedzialnych za odporność humoralną (realizowaną przez wyspecjalizowane komórki plazmatyczne zdolne do produkcji przeciwciał swoistych – specyficznych tylko wobec określonego antygenu) pełni u człowieka szpik kostny. Śledziona pełni rolę eliminującą krwinki czerwone, białe i płytkowe, które są uszkodzone lub osiągnęły swój kres życia, oraz jest miejscem produkcji przeciwciał, zwłaszcza przeciwko bakteriom posiadających otoczki.
Zasadniczą strukturą spełniającą funkcje układu chłonnego jest grudka chłonna (występująca w węzłach chłonnych i innych narządach limfatycznych). W części centralnej grudki chłonnej znajduje się ośrodek rozmnażania grudki chłonnej, w którym limfocyty B po zetknięciu się z antygenem, na który są przygotowane zareagować, ulegają przemianie i podziałom komórkowym, a następnie przekształcaniu do komórek plazmatycznych wydzielających swoiste przeciwciała. W otoczeniu grudki chłonnej znajduje się strefa płaszcza, a najbardziej zewnętrznie – strefa brzeżna. W strefie płaszcza znajdują się drobne limfocyty pamięci, które w przeszłości powstały w wyniku przemiany komórki wyjściowej pod wpływem spotkania antygenu.