Czym jest szpiczak mnogi?
Szpiczak mnogi (inaczej szpiczak plazmocytowy) jest nadmiernym i nieprawidłowym mnożeniem się nieprawidłowych plazmocytów (komórek plazmatycznych, czyli komórek produkujących przeciwciała – cząsteczki, które odpowiadają za odporność organizmu człowieka), najczęściej zlokalizowanym w kościach płaskich. Ta grupa schorzeń obejmuje też białaczkę plazmocytarną oraz pozaszpikową postać szpiczaka. Do chorób nowotworowych należących do nieprawidłowości plazmocytów zalicza się również pierwotną układową amyloidozę łańcuchów lekkich, zespół POEMS oraz makroglobulinemię Waldenströma.
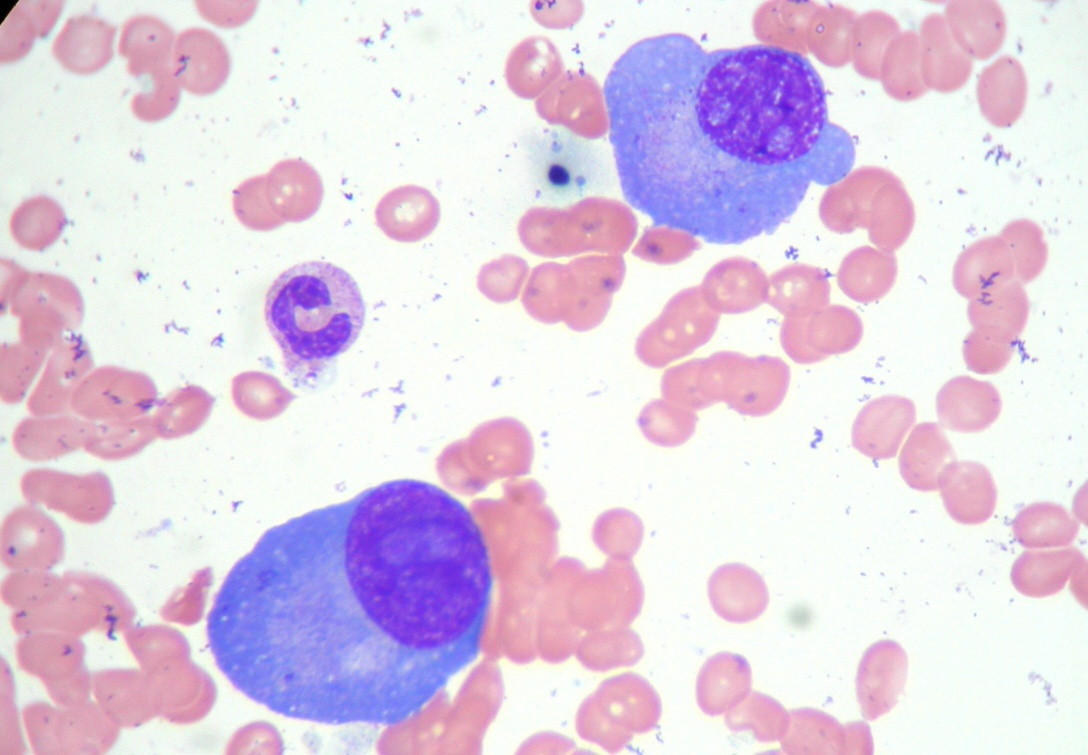
Komórki szpiczaka (fot. 1) produkują nieprawidłowe białko, które jest przeciwciałem lub też fragmentem przeciwciała (łańcuchy lekkie kappa lub lambda) (15-20% przypadków szpiczaka).
W 2009 roku szpiczak stanowił w Polsce drugą pod względem liczby nowych zarejestrowanych przypadków chorobę nowotworową układu chłonnego u dorosłych. Zachorowalność wynosi 1 do 8 przypadków na 100 tysięcy mieszkańców rocznie i jest większa w krajach zachodniej półkuli. Szpiczak występuje częściej u mężczyzn niż u kobiet oraz dwukrotnie częściej u osób rasy czarnej niż kaukaskiej. Nie występuje u dzieci i niezwykle rzadko rozpoznaje się go u osób poniżej 30. roku życia. Większość przypadków (90%) odnotowuje się u osób powyżej 50. roku życia.
Fot. 1. Komórki plazmatyczne w szpiku kostnym, powiększenie 1000×, barwienie metodą Wrighta (fot. dr Marta Szostek).]